
DETCI¶
Performs configuration interaction (CI) computations of various types, including restricted-active-space (RAS) CI, full CI, the CI component of multi-configuration self-consistent-field (MCSCF) and complete-active-space self-consistent-field (CASSCF) computations, and arbitrary-order perturbation theory and arbitrary-order coupled-cluster computations for small molecules.
General Options¶
AVG_STATES¶
Array giving the root numbers of the states to average in a state-averaged procedure such as SA-CASSCF. Root numbering starts from 1.
- Type: array
- Default: No Default
AVG_WEIGHTS¶
Array giving the weights for each state in a state-averaged procedure
- Type: array
- Default: No Default
A_RAS3_MAX¶
maximum number of alpha electrons in RAS III
- Type: integer
- Default: -1
B_RAS3_MAX¶
maximum number of beta electrons in RAS III
- Type: integer
- Default: -1
CIBLKS_PRINT¶
Do print a summary of the CI blocks?
- Type: boolean
- Default: false
CI_MAXITER¶
Maximum number of iterations to diagonalize the Hamiltonian
- Type: integer
- Default: 24
CI_NUM_THREADS¶
Number of threads for DETCI.
- Type: integer
- Default: 1
DETCI_FREEZE_CORE¶
Do freeze core orbitals?
- Type: boolean
- Default: true
E_CONVERGENCE¶
Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-6
ICORE¶
Specifies how to handle buffering of CI vectors. A value of 0 makes the program perform I/O one RAS subblock at a time; 1 uses entire CI vectors at a time; and 2 uses one irrep block at a time. Values of 0 or 2 cause some inefficiency in the I/O (requiring multiple reads of the C vector when constructing H in the iterative subspace if DIAG_METHOD = SEM), but require less core memory.
- Type: integer
- Default: 1
ISTOP¶
Do stop DETCI after string information is formed and before integrals are read?
- Type: boolean
- Default: false
NUM_DETS_PRINT¶
Number of important determinants to print
- Type: integer
- Default: 20
R_CONVERGENCE¶
Convergence criterion for CI residual vector in the Davidson algorithm (RMS error). The default is 1e-4 for energies and 1e-7 for gradients.
- Type: conv double
- Default: 1e-4
S¶
The value of the spin quantum number
is given by this option. The default is determined by the value of the multiplicity. This is used for two things: (1) determining the phase of the redundant half of the CI vector when the
component is used (i.e., MS0 =
TRUE), and (2) making sure the guess vector has the desired value of(if CALC_S_SQUARED is
TRUEand ICORE =1).
- Type: double
- Default: 0.0
VAL_EX_LEVEL¶
In a RAS CI, this is the additional excitation level for allowing electrons out of RAS I into RAS II. The maximum number of holes in RAS I is therefore EX_LEVEL + VAL_EX_LEVEL.
- Type: integer
- Default: 0
Diagonalization Methods¶
DIAG_METHOD¶
This specifies which method is to be used in diagonalizing the Hamiltonian. The valid options are:
RSP, to form the entire H matrix and diagonalize using libciomr to obtain all eigenvalues (n.b. requires HUGE memory);OLSEN, to use Olsen’s preconditioned inverse subspace method (1990);MITRUSHENKOV, to use a 2x2 Olsen/Davidson method; andDAVIDSON(orSEM) to use Liu’s Simultaneous Expansion Method, which is identical to the Davidson method if only one root is to be found. There also exists a SEM debugging mode,SEMTEST. TheSEMmethod is the most robust, but it also requiresCI vectors on disk, where
is the maximum number of iterations and
is the number of roots.
- Type: string
- Possible Values: RSP, OLSEN, MITRUSHENKOV, DAVIDSON, SEM, SEMTEST
- Default: SEM
LSE¶
Do use least-squares extrapolation in iterative solution of CI vector?
- Type: boolean
- Default: false
LSE_COLLAPSE¶
Number of iterations between least-squares extrapolations
- Type: integer
- Default: 3
LSE_TOLERANCE¶
Minimum converged energy for least-squares extrapolation to be performed
- Type: conv double
- Default: 3
PRECONDITIONER¶
This specifies the type of preconditioner to use in the selected diagonalization method. The valid options are:
DAVIDSONwhich approximates the Hamiltonian matrix by the diagonal elements;H0BLOCK_INVwhich uses an exact Hamiltonian of H0_BLOCKSIZE and explicitly inverts it;GEN_DAVIDSONwhich does a spectral decomposition of H0BLOCK;ITER_INVusing an iterative approach to obtain the correction vector of H0BLOCK. TheH0BLOCK_INV,GEN_DAVIDSON, andITER_INVapproaches are all formally equivalent but theITER_INVis less computationally expensive. Default isDAVIDSON.
- Type: string
- Possible Values: LANCZOS, DAVIDSON, GEN_DAVIDSON, H0BLOCK, H0BLOCK_INV, ITER_INV, H0BLOCK_COUPLING, EVANGELISTI
- Default: DAVIDSON
Density Matrices¶
NAT_ORBS¶
Build natural orbitals? The orbtials will be reordered by occuption number.
- Type: boolean
- Default: false
OPDM¶
Do compute one-particle density matrix if not otherwise required?
- Type: boolean
- Default: false
OPDM_AVG¶
Do average the OPDM over several roots in order to obtain a state-average one-particle density matrix? This density matrix can be diagonalized to obtain the CI natural orbitals.
- Type: boolean
- Default: false
OPDM_PRINT¶
Do print the one-particle density matrix for each root?
- Type: boolean
- Default: false
TDM¶
Do compute the transition density? Note: only transition densities between roots of the same symmetry will be evaluated. DETCI does not compute states of different irreps within the same computation; to do this, lower the symmetry of the computation.
- Type: boolean
- Default: false
TPDM_PRINT¶
Do print the two-particle density matrix? (Warning: large tensor)
- Type: boolean
- Default: false
Root Following¶
FOLLOW_ROOT¶
The root to write out the two-particle density matrix for (the one-particle density matrices are written for all roots). Useful for a state-specific CASSCF or CI optimization on an excited state.
- Type: integer
- Default: 1
Guess Vectors¶
File Handling¶
COLLAPSE_SIZE¶
Gives the number of vectors to retain when the Davidson subspace is collapsed (see MAX_NUM_VECS . If greater than one, the collapsed subspace retains the best estimate of the CI vector for the previous n iterations. Defaults to 1.
- Type: integer
- Default: 1
MAX_NUM_VECS¶
Maximum number of Davidson subspace vectors which can be held on disk for the CI coefficient and sigma vectors. (There is one H(diag) vector and the number of D vectors is equal to the number of roots). When the number of vectors on disk reaches the value of MAX_NUM_VECS, the Davidson subspace will be collapsed to COLLAPSE_SIZE vectors for each root. This is very helpful for saving disk space. Defaults to CI_MAXITER * NUM_ROOTS + NUM_INIT_VECS
- Type: integer
- Default: 0
General-Order Perturbation Theory¶
MPN¶
Do compute the MPn series out to kth order where k is determined by MAX_NUM_VECS ? For open-shell systems REFERENCE is ROHF, WFN is ZAPTN), DETCI will compute the ZAPTn series. GUESS_VECTOR must be set to UNIT, HD_OTF must be set to TRUE, and HD_AVG must be set to orb_ener; these should happen by default for MPN = TRUE.
- Type: boolean
- Default: false
General-Order Coupled-Cluster¶
CC_A_RAS3_MAX¶
maximum number of alpha electrons in RAS III, for CC
- Type: integer
- Default: -1
CC_B_RAS3_MAX¶
maximum number of beta electrons in RAS III, for CC
- Type: integer
- Default: -1
CC_EX_LEVEL¶
The CC excitation level
- Type: integer
- Default: 2
CC_RAS34_MAX¶
maximum number of electrons in RAS III + IV, for CC
- Type: integer
- Default: -1
CC_RAS3_MAX¶
maximum number of electrons in RAS III, for CC
- Type: integer
- Default: -1
CC_RAS4_MAX¶
maximum number of electrons in RAS IV, for CC
- Type: integer
- Default: -1
CC_VAL_EX_LEVEL¶
The CC valence excitation level
- Type: integer
- Default: 0
CC_VECS_READ¶
Do import a CC vector from disk?
- Type: boolean
- Default: false
CC_VECS_WRITE¶
Do export a CC vector to disk?
- Type: boolean
- Default: false
DIIS_FREQ¶
How often to do a DIIS extrapolation. 1 means do DIIS every iteration, 2 is every other iteration, etc.
- Type: integer
- Default: 1
DIIS_MAX_VECS¶
Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 5
DIIS_MIN_VECS¶
Minimum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 2
DIIS_START_ITER¶
Iteration at which to start using DIIS
- Type: integer
- Default: 1
NUM_AMPS_PRINT¶
Number of important CC amplitudes per excitation level to print. CC analog to NUM_DETS_PRINT
- Type: integer
- Default: 10
MCSCF¶
DF_BASIS_MCSCF¶
Auxiliary basis set for MCSCF density fitted ERI computations. This only effects the “Q” matrix in Helgaker’s language. Defaults to a JKFIT basis.
- Type: string
- Possible Values: basis string
- Default: No Default
MCSCF_DIIS_FREQ¶
How often to do a DIIS extrapolation
- Type: integer
- Default: 1
MCSCF_DIIS_MAX_VECS¶
Maximum number of DIIS vectors
- Type: integer
- Default: 8
MCSCF_DIIS_START¶
Iteration to turn on DIIS
- Type: integer
- Default: 3
MCSCF_E_CONVERGENCE¶
Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-7
MCSCF_MAXITER¶
Maximum number MCSCF of iterations
- Type: integer
- Default: 30
MCSCF_MAX_ROT¶
Maximum value in the rotation matrix. If a value is greater than this number all values are scaled.
- Type: double
- Default: 0.5
MCSCF_R_CONVERGENCE¶
Convergence criterion for the RMS of the orbital gradient
- Type: conv double
- Default: 1e-4
Expert General Options¶
EX_ALLOW¶
An array of length EX_LEVEL specifying whether each excitation type (S,D,T, etc.) is allowed (1 is allowed, 0 is disallowed). Used to specify non-standard CI spaces such as CIST.
- Type: array
- Default: No Default
R4S¶
Do restrict strings with
in RAS IV? Useful to reduce the number of strings required if MIXED4=true, as in a split-virutal CISD[TQ] computation. If more than one electron is in RAS IV, then the holes in RAS I cannot exceed the number of particles in RAS III + RAS IV (i.e., EX_LEVEL , or else the string is discarded.
- Type: boolean
- Default: false
SF_RESTRICT¶
Do eliminate determinants not valid for spin-complete spin-flip CI’s? [see J. S. Sears et al, J. Chem. Phys. 118, 9084-9094 (2003)]
- Type: boolean
- Default: false
SIGMA_OVERLAP¶
Do print the sigma overlap matrix? Not generally useful.
- Type: boolean
- Default: false
Expert Diagonalization Methods¶
H0_BLOCKSIZE¶
This parameter specifies the size of the H0 block of the Hamiltonian which is solved exactly. The n determinants with the lowest SCF energy are selected, and a submatrix of the Hamiltonian is formed using these determinants. This submatrix is used to accelerate convergence of the CI iterations in the OLSEN and MITRUSHENKOV iteration schemes, and also to find a good starting guess for the SEM method if GUESS_VECTOR is
H0_BLOCK. Defaults to 1000. Note that the program may change the given size for Ms=0 cases MS0 is TRUE) if it determines that the H0 block includes only one member of a pair of determinants related by time reversal symmetry. For very small block sizes, this could conceivably eliminate the entire H0 block; the program should print warnings if this occurs.
- Type: integer
- Default: 1000
H0_BLOCK_COUPLING¶
Do use coupling block in preconditioner?
- Type: boolean
- Default: false
H0_BLOCK_COUPLING_SIZE¶
Parameters which specifies the size of the coupling block within the generalized davidson preconditioner.
- Type: integer
- Default: 0
H0_GUESS_SIZE¶
size of H0 block for initial guess
- Type: integer
- Default: 1000
HD_AVG¶
How to average H diag energies over spin coupling sets.
HD_EXACTuses the exact diagonal energies which results in expansion vectors which break spin symmetry.HD_KAVEaverages the diagonal energies over a spin-coupling set yielding spin pure expansion vectors.ORB_ENERemploys the sum of orbital energy approximation giving spin pure expansion vectors but usually doubles the number of Davidson iterations.EVANGELISTIuses the sums and differences of orbital energies with the SCF reference energy to produce spin pure expansion vectors.LEININGERapproximation which subtracts the one-electron contribution from the orbital energies, multiplies by 0.5, and adds the one-electron contribution back in, producing spin pure expansion vectors and developed by Matt Leininger and works as well asEVANGELISTI.
- Type: string
- Possible Values: EVANGELISTI, HD_EXACT, HD_KAVE, ORB_ENER, LEININGER, Z_KAVE
- Default: EVANGELISTI
Expert Density Matrices¶
Expert Root Following¶
FOLLOW_VECTOR¶
In following a particular root (see FOLLOW_ROOT , sometimes the root number changes. To follow a root of a particular character, one can specify a list of determinants and their coefficients, and the code will follow the root with the closest overlap. The user specifies arrays containing the absolute alpha string indices (A_i below), absolute beta indices (B_i below), and CI coefficients (C_i below) to form the desired vector. The format is FOLLOW_VECTOR = [ [[A_1, B_1], C_1], [[A_2, B_2], C_2], ...].
- Type: array
- Default: No Default
Expert Guess Vectors¶
CI_FILE_START¶
What file do we start at for hd/c/s/d CIvects? Should be 50 for normal CI calculations and 54 if we are going to do a second monomer.
- Type: integer
- Default: 50
FILTER_GUESS¶
Do invoke the FILTER_GUESS options that are used to filter out some trial vectors which may not have the appropriate phase convention between two determinants? This is useful to remove, e.g., delta states when a sigma state is desired. The user inputs two determinants (by giving the absolute alpha string number and beta string number for each), and also the desired phase between these two determinants for guesses which are to be kept. FILTER_GUESS = TRUE turns on the filtering routine. Requires additional keywords FILTER_GUESS_DET1 FILTER_GUESS_DET2 and FILTER_GUESS_SIGN
- Type: boolean
- Default: false
FILTER_GUESS_DET1¶
Array specifying the absolute alpha string number and beta string number for the first determinant in the filter procedure. (See FILTER_GUESS .
- Type: array
- Default: No Default
FILTER_GUESS_DET2¶
Array specifying the absolute alpha string number and beta string number for the second determinant in the filter procedure. (See FILTER_GUESS .
- Type: array
- Default: No Default
FILTER_GUESS_SIGN¶
The required phase (1 or -1) between the two determinants specified by FILTER_GUESS_DET1 and FILTER_GUESS_DET2
- Type: integer
- Default: 1
FILTER_ZERO_DET¶
If present, the code will try to filter out a particular determinant by setting its CI coefficient to zero. FILTER_ZERO_DET = [alphastr, betastr] specifies the absolute alpha and beta string numbers of the target determinant. This could be useful for trying to exclude states that have a nonzero CI coefficient for the given determinant. However, this option was experimental and may not be effective.
- Type: array
- Default: No Default
GUESS_VECTOR¶
Guess vector type. Accepted values are
UNITfor a unit vector guess NUM_ROOTS and NUM_INIT_VECS must both be 1);H0_BLOCKto use eigenvectors from the H0 BLOCK submatrix (default);DFILEto use NUM_ROOTS previously converged vectors in the D file;
- Type: string
- Possible Values: UNIT, H0_BLOCK, DFILE
- Default: H0_BLOCK
NUM_INIT_VECS¶
The number of initial vectors to use in the CI iterative procedure. Defaults to the number of roots.
- Type: integer
- Default: 0
REFERENCE_SYM¶
Irrep for CI vectors; -1 = find automatically. This option allows the user to look for CI vectors of a different irrep than the reference. This probably only makes sense for Full CI, and it would probably not work with unit vector guesses. Numbering starts from zero for the totally-symmetric irrep.
- Type: integer
- Default: -1
Expert File Handling¶
Expert General-Order Perturbation Theory¶
MPN_ORDER_SAVE¶
If 0, save the MPn energy; if 1, save the MP(2n-1) energy (if available from MPN_WIGNER = true); if 2, save the MP(2n-2) energy (if available from MPN_WIGNER = true).
- Type: integer
- Default: 0
MPN_SCHMIDT¶
Do employ an orthonormal vector space rather than storing the kth order wavefunction?
- Type: boolean
- Default: false
MPN_WIGNER¶
Do use Wigner formulas in the
series?
- Type: boolean
- Default: true
PERTURB_MAGNITUDE¶
The magnitude of perturbation
in
- Type: double
- Default: 1.0
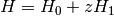
Expert General-Order Coupled-Cluster¶
CC_FIX_EXTERNAL¶
Do fix amplitudes involving RAS I or RAS IV? Useful in mixed MP2-CC methods.
- Type: boolean
- Default: false
CC_FIX_EXTERNAL_MIN¶
Number of external indices before amplitude gets fixed by CC_FIX_EXTERNAL Experimental.
- Type: integer
- Default: 1
CC_MACRO¶
CC_MACRO = [ [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], ... ] Optional additional restrictions on allowed exictations in coupled-cluster computations, based on macroconfiguration selection. For each sub-array, [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], eliminate cluster amplitudes in which: [the excitation level (holes in I + II) is equal to ex_lvl] AND [there are more than max_holes_I holes in RAS I, there are more than max_parts_IV particles in RAS IV, OR there are more than max_I+IV quasiparticles in RAS I + RAS IV].
- Type: array
- Default: No Default
CC_MIXED¶
Do ignore block if num holes in RAS I and II is
cc_ex_lvl and if any indices correspond to RAS I or IV (i.e., include only all-active higher excitations)?
- Type: boolean
- Default: true
CC_UPDATE_EPS¶
Do update T amplitudes with orbital eigenvalues? (Usually would do this). Not doing this is experimental.
- Type: boolean
- Default: true
CC_VARIATIONAL¶
Do use variational energy expression in CC computation? Experimental.
- Type: boolean
- Default: false
Expert Alternative Algorithms¶
BENDAZZOLI¶
Do use some routines based on the papers of Bendazzoli et al. to calculate sigma? Seems to be slower and not worthwhile; may disappear eventually. Works only for full CI and I don’t remember if I could see how their clever scheme might be extended to RAS in general.
- Type: boolean
- Default: false
FCI_STRINGS¶
Do store strings specifically for FCI? (Defaults to TRUE for FCI.)
- Type: boolean
- Default: false
REPL_OTF¶
Do string replacements on the fly in DETCI? Can save a gigantic amount of memory (especially for truncated CI’s) but is somewhat flaky and hasn’t been tested for a while. It may work only works for certain classes of RAS calculations. The current code is very slow with this option turned on.
- Type: boolean
- Default: false
Expert MCSCF¶
MCSCF_ALGORITHM¶
Convergence algorithm to utilize. This is a flag for the future.
- Type: string
- Possible Values: ONE_STEP, TWO_STEP
- Default: TWO_STEP
MCSCF_SO¶
Do second-order orbital-orbital MCSCF. Without one-step this typically slows the overall computation considerably
- Type: boolean
- Default: false
MCSCF_SO_START_E¶
Start second-order orbital-orbital MCSCF based on energy convergence
- Type: double
- Default: 1e-3
MCSCF_SO_START_GRAD¶
Start second-order orbital-orbital MCSCF based on RMS of orbital gradient
- Type: double
- Default: 1e-3