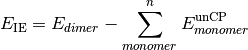PSI Variables by Alpha¶
Note
Lowercase letters in PSI variable names represent portions of the variable name that vary by root number, calculation order, etc. See text for fuller description.
- (T) CORRECTION ENERGY¶
The coupled-cluster perturbative triples correction [H].
- AAA (T) CORRECTION ENERGY¶
- AAB (T) CORRECTION ENERGY¶
- ABB (T) CORRECTION ENERGY¶
- BBB (T) CORRECTION ENERGY¶
Components of the coupled-cluster perturbative triples correction [H].
- BRUECKNER CONVERGED¶
Value 1 (0) when the Brueckner orbitals have (have not) converged.
- CBS TOTAL ENERGY¶
- CBS CORRELATION ENERGY¶
- CBS REFERENCE ENERGY¶
The total electronic energy [H] and its breakdown into reference total energy [H] and correlation correction components [H] for the compound method requested through cbs().
- CC ROOT n TOTAL ENERGY¶
The total electronic energy [H] for the requested coupled cluster level of theory and root n (numbering starts at GS = 0).
- CC2 TOTAL ENERGY¶
- CC2 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the CC2 level of theory.
- CC3 TOTAL ENERGY¶
- CC3 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the CC3 level of theory.
- CCSD TOTAL ENERGY¶
- CCSD CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the CCSD level of theory.
- CCSD(T) TOTAL ENERGY¶
- CCSD(T) CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the CCSD(T) level of theory.
- CI DIPOLE X¶
- CI DIPOLE Y¶
- CI DIPOLE Z¶
The three components of the dipole [Debye] for the requested configuration interaction level of theory and root.
- CI QUADRUPOLE XX¶
- CI QUADRUPOLE XY¶
- CI QUADRUPOLE XZ¶
- CI QUADRUPOLE YY¶
- CI QUADRUPOLE YZ¶
- CI QUADRUPOLE ZZ¶
The six components of the quadrupole [Debye Ang] for the requested configuration interaction level of theory and root.
- CI ROOT n -> ROOT m DIPOLE X¶
- CI ROOT n -> ROOT m DIPOLE Y¶
- CI ROOT n -> ROOT m DIPOLE Z¶
The three components of the transition dipole [Debye] between roots n and m for the requested configuration interaction level of theory.
- CI ROOT n -> ROOT m QUADRUPOLE XX¶
- CI ROOT n -> ROOT m QUADRUPOLE XY¶
- CI ROOT n -> ROOT m QUADRUPOLE XZ¶
- CI ROOT n -> ROOT m QUADRUPOLE YY¶
- CI ROOT n -> ROOT m QUADRUPOLE YZ¶
- CI ROOT n -> ROOT m QUADRUPOLE ZZ¶
The three components of the transition quadrupole [Debye Ang] between roots n and m for the requested configuration interaction level of theory.
- CI ROOT n DIPOLE X¶
- CI ROOT n DIPOLE Y¶
- CI ROOT n DIPOLE Z¶
The three components of the dipole [Debye] for the requested configuration interaction level of theory and root n.
- CI ROOT n QUADRUPOLE XX¶
- CI ROOT n QUADRUPOLE XY¶
- CI ROOT n QUADRUPOLE XZ¶
- CI ROOT n QUADRUPOLE YY¶
- CI ROOT n QUADRUPOLE YZ¶
- CI ROOT n QUADRUPOLE ZZ¶
The six components of the quadrupole [Debye Ang] for the requested configuration interaction level of theory and root n.
- CI ROOT n TOTAL ENERGY¶
- CI ROOT n CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the requested configuration interaction level of theory and root n (numbering starts at 1).
- CI STATE-AVERAGED TOTAL ENERGY¶
- CI STATE-AVERAGED CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for state-averaged CI/CASSCF levels of theory.
- CI TOTAL ENERGY¶
- CI CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the requested configuration interaction level of theory and root.
- CISD TOTAL ENERGY¶
- CISD CORRELATION ENERGY¶
- CISDT TOTAL ENERGY¶
- CISDT CORRELATION ENERGY¶
- CISDTQ CORRELATION ENERGY¶
- CISDTQ TOTAL ENERGY¶
- CIn CORRELATION ENERGY¶
- CIn TOTAL ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the labeled configuration interaction level of theory and root. n is CI order for n > 4.
- CP-CORRECTED 2-BODY INTERACTION ENERGY¶
The interaction energy [H] considering only two-body interactions, computed with counterpoise correction. Related variable UNCP-CORRECTED 2-BODY INTERACTION ENERGY.
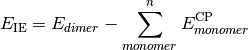
- CURRENT CORRELATION ENERGY¶
The correlation energy [H] corresponding to the CURRENT ENERGY variable.
- CURRENT ENERGY¶
The total electronic energy [H] of the most recent stage of a calculation (frequently overwritten). This is the quantity tracked by the geometry optimizer.
- CURRENT REFERENCE ENERGY¶
The total electronic energy [H] of the reference stage corresponding to the CURRENT ENERGY variable.
- db_name DATABASE MEAN ABSOLUTE DEVIATION¶
The mean absolute deviation [kcal mol-1] of the requested method name from the stored reference values for the requested reactions in database db_name. If no reference is available, this will be a large and nonsensical value.
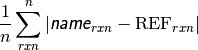
- db_name DATABASE MEAN SIGNED DEVIATION¶
The mean deviation [kcal mol-1] of the requested method name from the stored reference values for the requested reactions in database db_name. If no reference is available, this will be a large and nonsensical value.
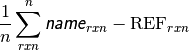
- db_name DATABASE ROOT-MEAN-SQUARE SIGNED DEVIATION¶
The rms deviation [kcal mol-1] of the requested method name from the stored reference values for the requested reactions in database db_name. If no reference is available, this will be a large and nonsensical value.
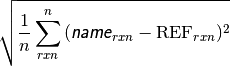
- DF-MP2 TOTAL ENERGY¶
- DF-MP2 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the density-fitted MP2 level of theory.
- DFT FUNCTIONAL ENERGY¶
The functional energy contribution [H] to the total SCF energy (DFT only). Quantity
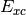 in Eq. (2).
in Eq. (2).
- DFT FUNCTIONAL TOTAL ENERGY¶
The total electronic energy [H] for the underlying functional of the requested DFT method, without any dispersion correction, the first four terms in Eq. (2). When the requested method includes a dispersion correction, this quantity is
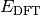 in Eq. (1).
Otherwise, quantity equal to DFT TOTAL ENERGY
and SCF TOTAL ENERGY.
in Eq. (1).
Otherwise, quantity equal to DFT TOTAL ENERGY
and SCF TOTAL ENERGY.
- DFT TOTAL ENERGY¶
The total electronic energy [H] for the requested DFT method,
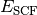 in Eq. (2).
When the method includes a dispersion correction, this quantity
is
in Eq. (2).
When the method includes a dispersion correction, this quantity
is 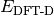 in Eq. (1).
in Eq. (1).
- DISPERSION CORRECTION ENERGY¶
The dispersion correction [H] appended to an underlying functional when a DFT-D method is requested. Quantity
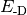 in Eqs. (1) and (2).
in Eqs. (1) and (2).(1)
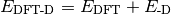
- FCI TOTAL ENERGY¶
- FCI CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the full configuration interaction level of theory.
- LCC2 (+LMP2) TOTAL ENERGY¶
The total electronic energy [H] for the local CC2 level of theory.
- LCCSD (+LMP2) TOTAL ENERGY¶
The total electronic energy [H] for the local CCSD level of theory.
- MP2 TOTAL ENERGY¶
- MP2 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the MP2 level of theory.
- MP2.5 TOTAL ENERGY¶
- MP2.5 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the MP2.5 level of theory.
- MP3 TOTAL ENERGY¶
- MP3 CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the MP3 level of theory.
- MPn TOTAL ENERGY¶
- MPn CORRELATION ENERGY¶
The total electronic energy [H] and correlation energy component [H] for the labeled Möller–Plesset perturbation theory level. n is MP perturbation order.
- NUCLEAR REPULSION ENERGY¶
The nuclear repulsion energy contribution [H] to the total SCF energy. Quantity
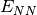 in Eq. (2).
in Eq. (2).
- ONE-ELECTRON ENERGY¶
The one-electron energy contribution [H] to the total SCF energy. Quantity
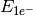 in Eq. (2).
in Eq. (2).
- SAPT DISP ENERGY¶
- SAPT ELST ENERGY¶
- SAPT EXCH ENERGY¶
- SAPT IND ENERGY¶
Respectively, the dispersion, electrostatics, exchange, and induction components of the total electronic interaction energy [H] for the the requested SAPT level of theory. The sum of these four components yields SAPT ENERGY.
- SAPT ENERGY¶
The total electronic interaction energy [H] for the requested SAPT level of theory.
- SAPT SAPT0 ENERGY¶
- SAPT SAPT2 ENERGY¶
- SAPT SAPT2+ ENERGY¶
- SAPT SAPT2+(3) ENERGY¶
- SAPT SAPT2+3 ENERGY¶
The total electronic interaction energy [H] for the labeled SAPT level of theory.
- SCF TOTAL ENERGY¶
The total electronic energy [H] of the SCF stage of the calculation. The CORRELATION ENERGY variables from subsequent stages of a calculation are often the corresponding TOTAL ENERGY variables less this quantity. Constructed from Eq. (2).
(2)
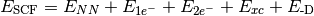
- TWO-ELECTRON ENERGY¶
The two-electron energy contribution [H] to the total SCF energy. Quantity
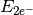 in Eq. (2).
in Eq. (2).
- UNCP-CORRECTED 2-BODY INTERACTION ENERGY¶
The interaction energy [H] considering only two-body interactions, computed without counterpoise correction. Related variable CP-CORRECTED 2-BODY INTERACTION ENERGY.